
In this review, we exposed the experimental evidences, related to the cGMP signals, linked to the muscarinic activation of airway smooth muscle (ASM), an essential element in the bronchoconstriction and the remodelling described in asthma and COPD. During this muscarinic activation, two cGMP signals are observed, at 20-s and 60-s, being products of two different muscarinic receptors (mAChRs) associated with two distinctive guanylylcyclases. Thus, the 20-s-cGMP signal involves an M2mAChR inducing a massive and transient α1β1- NO-sGC translocation from cytoplasm to plasma membranes. Moreover, the 60-s-cGMP signal is associated with a novel G-protein coupled NPR-GC-B. This nano-machine is regulated by MAChRs, in an opposite way, by an M3mAChR coupled to Gq16 to activate NPR-GC-B and an M2mAChR coupled to Go/1 to inhibit this NPR-GC-B. Moreover, M3mAChRs is desensitizated by dimerization induced by PKG-II phosphorylation. This phosphorylated-M3mAChR binds muscarinic antagonists rather than muscarinic agonists implying receptor desensitization. Recently, we described a novel PDE1A coupled to M2mAChR to finish these cGMP signal transduction cascades. In ASM cells from rats, M2mAChR displays a novel anti-mitogenic effects suggesting that a possible imbalance between these two M2mAChR/M3mAChR signal cascades can contribute to airway hyperreactivity and the abnormal proliferative ASM responses present in asthma and COPD. In asthma experimental model as OVA-sensitized rats, the muscarinic activation of these ASMC exhibited a dysfunction of these cascades involving two M2mAChR/M3mAChR associated with two distinctive cGMP generators, which has been implied in asthma and COPD, opening ways for the development of novel drugs to treat these human diseases.
Key words: cGMP; M2mAChR; M3mAChR; PKG-II; NPR-GC-B; NOs GC.
En esta revisión se presentan evidencias experimentales que relacionan al GMPc con la activación muscarínica del músculo liso de las vías aéreas, elemento esencial en la broncoconstricción y en la remodelación descrita tanto en el asma bronquial como en la enfermedad obstructiva crónica (COPD).
En efecto, durante la activación muscarínica se observan dos señales del GMPc una a los 20 segundos y otra a los 60 segundos, las cuales son el producto de 2 diferentes tipos de receptores muscarínicos asociados con dos diferentes guanililciclasas.
Esta nanomaquinaria está regulada en forma opuesta por receptores muscarínicos , en forma tal que hay un receptor muscarínico tipo M3 acoplado a una proteína G Gq16 para activar al complejo NPR-GC-B y un receptor muscarínico de tipo M2 acoplado a una Go/1 para inhibir este complejo NPR-GC-B.
Aún más, el receptor M3 es desensibilizado por dimerización inducida por fosforilación a través de una PKGII., además una nueva fosfodiesterasa PDE1A acoplada al receptor muscarínico M2 con lo cual se finaliza esta cascada de transducción de señales inducidas por el GMPc. Ladisfunción de estas cascadas que involucran a los M2 y M3, implicadas en el asma y en COPD, pudieran ser objeto del uso de nuevas drogas en estas dos patologías humanas.
Palabras clave: Guanosín monofosfato cíclico (cGMP); Receptor muscarínico tipo 2 (M2mAChR); Receptor muscarínico tipo 3 (M3mAChR); Proteín quinasa tipo II (PKG-II); una guanil-ciclasa tipo B sensible al Péptido natriurético (NPR-GC-B); Guanil-ciclasa sensible a óxido nitrico (NO)
The airway smooth muscle tone is mainly regulated by the dominant neuronal pathway under the parasympathetic nervous system control. Acetylcholine (Ach) is released under cholinergic stimulation being the predominant parasympathetic neurotransmitter; acting as an autocrine or paracrine hormone in the airways. The role of Ach in the regulation of bronchomotor tone (bronchoconstriction) and mucus secretion from airway submucosal glands in the respiratory tract is well established (Belmonte, 2005; Van der Velden & Hulsmann, 1999). Muscarinic activation of Airway smooth muscle (ASM) is of major importance to the physiological and patho-physiological actions of acetylcholine, which induces bronchoconstriction, airway smooth muscle thickening, may promoting airway inflammation and remodelling, including airway smooth muscle thickening and the modulation of cytokine and chemokine production by these cells inflammatory (Belmonte, 2005), which may contribute to the pathogenesis and pathophysiology of asthma as reported (Gosens et al., 2006; Racke & Matthiesen, 2004; Racke et al., 2006).
Neurogenic source of ACh has been related to the vagal nerve endings and the Non-neurogenic source of ACh is produced by the non-neuronal cells and tissues, particularly inflammatory cells and the airway epithelium as reported (Proskocil et al., 2004; Wessler & Kirkpatrick, 2001; Wessler & Kirkpatrick, 2008). Acetylcholine derived from the vagal nerve and from non-neuronal origins such as the airway epithelium acting on muscarinic receptors (mAChRs) anchored at the airway smooth muscle sarcolemma are involved in the generation of a number of signal transducing cascades allowing the activation of the smooth muscle machinery (Challiss et al., 1993). These mAChRs, which are members of the so called G protein-coupled receptors (GPCR) family, which are cell surface receptors, that activate intracellular responses by coupling G proteins to specific effectors (Oldham & Hamm, 2008; Kostenis et al., 1999). Molecular cloning studies have revealed the existence of five mammalian subtypes of muscarinic receptors (m1-m5) (Caulfield, 1993). Airway smooth muscle expresses mRNAs coding for both m2 and m3 receptors (Maeda et al., 1988) being identified using pharmacological ligand binding studies as a mixed population of M2 and M3 subtypes roughly in a 4:1 ratio (Lucchesi et al., 1990; Eglen et al., 1996). It is relevant that the M2 subtype is the most abundant mAchR in airway smooth muscle plasma membranes as reported (Lucchesi et al., 1990; Roffel et al., 1988; Misle et al., 1994; Misle et al., 2001).
M3mAChR represents a primary target of acetylcholine in the airways, which is involved in the regulation of bronchoconstriction (Meurs et al., 1988; Roffel et al., 1988; Eglen et al., 1996; Misle et al., 2001). Classically, M3mAChRs in ASM are coupled to phospholipase C (PLC)/protein kinase C (PKC) pathway via pertussis toxin (PTX)-insensitive G proteins of the Gq/11 family. The contractile response evoked by M3mAChRs stimulation is attributed to the formation of inositol trisphosphate (IP3), the subsequent release of Ca2+ from intracellular stores, the additional influx of extracellular calcium, and the Ca2+-sensitizing effect of PKC (Grandordy et al., 1986; Meurs et al., 1988; Roffel et. al 1990a; Roffel et al., 1990b).
The stimulation of M2 muscarinic receptors (M2mAChRs) in ASM inhibits adenylyl cyclase via activation of PTX-sensitive G proteins of the Gi/o family in (Jones et al., 1987; Sankary et al., 1988) and therefore M2mAChRs are thought to counteract relaxation and potentiate smooth muscle contraction (Fernandes et al., 1992). Experimental evidence has been provided that M2mAChRs participate directly in ASM contraction. In this sense, it has been shown that M2mAChRs stimulate Gi/o proteins to released βγ dimer, which inhibit the Large Conductance Ca2+- activated K+ Channel Activity (BK channels), Zhou et al., 2008. The inhibition of BK channel activity favors contraction of ASM and these BK channels are opposed to the M2mAChR-mediated depolarization and activation of calcium channels by restricting excitation–contraction coupling to more negative voltage ranges (Semenov et al., 2011).
In addition, the influence of M2mAChRs to modulate the relaxant effects of atrial natriuretic peptide (ANP) has been reported. Thus, the stimulation of M2mAChRs suppresses ANP-induced activation of particulate guanylyl cyclase via a PTX-sensitive G protein (Nakahara et al., 2002).
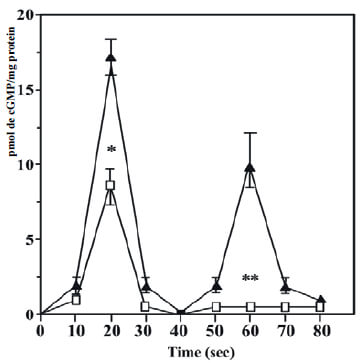
In this review, we address the generation of second messengers as cGMP, which is linked to some of the signal transducing cascades activated by M3mAChRs and M2mAChR, at intact Bovine ASM (BASM) strips. Thus, the muscarinic activation of airway smooth muscle (ASM) fragments associated with smooth muscle contraction, displays a kinetic behavior that involves the generation of two cGMP signals, at 20-s and 60-s (Guerra de González et al., 1999) as shown in Figure 1. Thus, the generation of these two signal cGMP are mediated by an M2mAChR coupled to PTX sensitive G-protein inducing the transient translocation of NO-sensitive soluble guanylylcyclases (NO-sGC) (Pláceres-Uray et al., 2010a) from ASM cytoplasm to the sarcolemma to produce the 20 s signal. In the other hand, an M3mAChR coupled to a Gq16 protein acting on the plasma membranespanning Natriuretic peptide Receptor Guanylyl Cyclases type B (NPR-GC-B) (Lippo de Bécemberg et al., 1995; Alfonzo et al., 1998a; Alfonzo et al., 1998b; Borges et al., 2001; Alfonzo et al., 2006; Bruges et al. 2007) to be responsible of the 60 s signal. In addition, both (NO-sGC) and (NPR-GC-B) were evaluated in a broken cell system as plasma membranes fractions isolated from Bovine ASM. These cGMP signal cascades were ended by two ways, one, this second messenger activates a negative feedback mechanism involving a PKG-II-dependent phosphorylation to specifically desensitize M3mAChR (Alfonzo et al., 2013; Alfonzo et al., 2015) the main receptor, involves in ASM contraction. Moreover, to degrade these cGMP signals, a novel mechanism of activation of a PDE1A-coupled to M2mAChR (Alfonzo et al., 2015) is discussed. To establish, the functional relevant of these M2mAChR/NO-sGC and M3mAChR/ NPR-GC-B cascades, an additional approach, using Airway Smooth Muscles Cells (ASMC) in culture was performed. This approach allows a new insight into the role of M2mAChR as anti-proliferative agent in ASMC (Pláceres-Uray et al., 2013). Furthermore, the functional roles of mAChRs linked GC systems (M2mAChR/NO-sGC and M3mAChR/NPR-GC-B) were evaluated in ASMC in culture obtained from an asthma experimental model using OVA-sensitized rats. In these experiments, an imbalance in these GC activities expressed as a depressed NO-sGC activity (Pláceres-Uray et al., 2010b) and an hyperstimulation of the M3mAChR/NPR-GC was found (Pláceres-Uray et al., 2011).
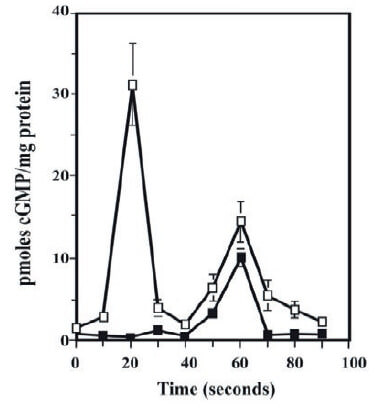
Until now, the muscarinic activation of BASM is unique biological system described that involves the generation of two cGMP signals as second messengers (Guerra de González et al., 1999; Borges et al., 2001). Interestingly, the 20-s cGMP signal is linked to the onset and the 60s cGMP signal is related to the “plateau” of the airway smooth muscle contraction (Guerra de González et al., 1999). This 20-s signal is also associated with the NO-sGC activity (Pláceres- Uray et al., 2010a) and the second, the 60-s signal, is linked to the activation of a membrane-bound NPR-GC (Lippo de Bécemberg et al., 1995; Alfonzo et al., 1998a;
Alfonzo et al., 1998b; Borges et al., 2001; Alfonzo et al., 2006; Bruges et al 2007), which has been previously characterized at BASM. The details of the activation of an NO-sGC, generating the early 20-s cGMP signal, presents in the plasma membrane of BASM, has been previously reported (Pláceres-Uray et al., 2010a) and further analyzed in this review.
The NO-sGCs are stimulated by a primary and beststudied endogenous activator, which is nitric oxide (NO). The NO-sGC is a heterodimeric hemoprotein formed by two different subunits, α- and β-subunits, which exist in four types (α1, α2, β1, and β2), each ones being the product of a separate gene (Sharina et al., 2000). Structurally, each subunit consists of N-terminal H-NOX domain, a central domain related to the dimerization, and a C-terminal consensus nucleotide catalytic cyclase domain (Wedel et al., 1995; Derbyshire & Marletta, 2012). For the formation of a catalytically active enzyme, both α- and β-subunits are required (Buechler et al., 1991). Although the α1β1 isoform is ubiquitous, the α2β1 isoform is less broadly distributed (Buechler et al., 1991; Wedel et al., 1995; Derbyshire & Marletta 2012). The best-characterized heterodimers are the α1/β1 and the α2/β1 isoforms (Wagner et al., 2005) being the first ones, relevant in ASM (Pláceres-Uray et al., 2010a). At molecular level, His-105 at the amino terminus of the β1 subunit of NO-sGC is the axial ligand of the penta-coordinated reduced iron center of heme, which is required for NO activation (Russwurn et al., 2013).
The first 20-s cGMP signal is a product of NO-sGC linked to M2mAChR coupled to G-proteins sensitive to PTX, which was established with several experimental approaches, as biochemical, pharmacological and molecular biology methods. This statement is supported by the following experimental evidences (Pláceres-Uray et al., 2010a). Thus, in intact ASM strips exposed, to muscarinic agonist (Cch), the isolated plasma membranes fractions from these Cch exposed ASM strips displayed increments in the GC activity at 20s and the 60s. Interestingly, at 20s, there is a peak of GC activity being stimulated in several fold by a NO releasing compound as SNP, but the 60s GC peak was not affected by SNP as shown in Figure 2. These GC activities increments at 20 and 60s coincide, with the two cGMP signals, reported at BASM strips, under agonist exposure (see Figure 1) (Guerra de González et al., 1999).
![Figure 2 Time-course of guanylyl cyclase activities in plasma membrane fractions isolated from BASM strips under muscarinic agonist exposure. Isolated BASM strips were incubated in the presence and absence of muscarinic agonist Cch for 70 s (Pláceres-Uray et al., 2010a). After each 10 s, the BASM strips were removed and immediately frozen in liquid N2 and processed to prepare plasma membranes fraction (González de Alfonzo et al., 1996). In these membrane fractions, GC assays as cGMP determinations were performed in duplicate as described (Lippo de Bécemberg, 1995). Empty symbols represent basal GC activities. Full symbols represent GC activities in the presence of 100 μM SNP. Basal (□), muscarinic agonist [Cch 1 x 10−5 M] (○), basal plus SNP (▲), and Cch + SNP (■). Each value is the mean of three different BASM strips. Taken from (Pláceres-Uray et al., 2010a).](2.jpg)
Additionally, in these plasma membranes isolated from BASM showed that the NO-sGC heterodimer involved is the α1β1. The Western blotting analysis with specific antibodies against all subunits of NO-sGC demonstrate that under muscarinic activation, the α1β1NO-sGC-heterodimer isoform is translocated from cytoplasm to plasma membranes of BASM are shown in Figure 3. The NO-sGC activities have been also described at plasma membrane from cardiomyocytes (Agulló et al., 2005) and neurons (Bidmon et al., 2006). Moreover, it has been reported that the α2β2 isoform of NO-sGCs at plasma membrane site, may provide a localized pool of cGMP (Agulló et al., 2005; Bellingham & Evans, 2007), which seems to be in similar trend postulated in this review.
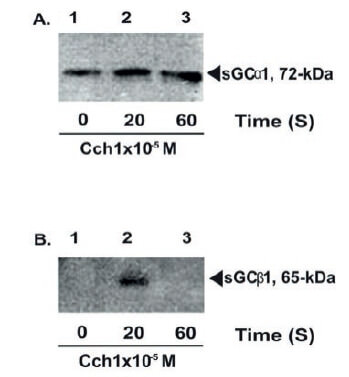
Furthermore, these experimental findings exhibited in Figure 3 revealed, an interesting fact, which involved the formation of the transient active α1β1 dimer. It can be seen, after muscarinic agonist exposure, that α1 subunits are present, all the time, at the plasma membrane, but, the β1 subunit is only present at 20s to form the active α1β1 dimer. The transient formation of α1β1 NO-sGC mechanism can explain the existence of the first 20s cGMP signal observed in intact BASM strips under muscarinic stimulation as shown in Figure 1.
These experiments were also supported by a series of additional reconstituted experiments using isolated plasma membranes fractions and concentrated cytosol fractions from Bovine ASM (Pláceres-Uray et al., 2010a). In these reconstituted experiments, there is a muscarinic agonist-dependent “incorporation” of NO-sGC to plasma membranes fragments being sensitive to PTX involving a Go/Gi heterotrimeric proteins as previously demonstrated (Pláceres-Uray et al., 2010a). Thus, the β1-sGC subunit of the α1β1 is translocated from cytoplasm to the inner face of the airway smooth muscle sarcolemma under muscarinic activation. Since the capability of this α1β1-sGC to migrate to plasma membranes under muscarinic activation, a purification procedure and further identification of this α1β1-sGC heterodimer was also performed (Pláceres-Uray et al., 2010a). In summary, all these experimental evidences support the rationale that first 20s cGMP signal is a product of a novel signaling cascade involving M2mAChR coupled to Go/i protein, which facilitates the transient formation of the α1β1-sGC isoform by migration of the β1 subunit from cytoplasm to the BASM sarcolemma. All these experimental evidences support a model for a novel signal transducing cascade integrates by the M2mAChR, Gi/o, and the α1β1 heterodimer of sGC as illustrated in Figure 4.
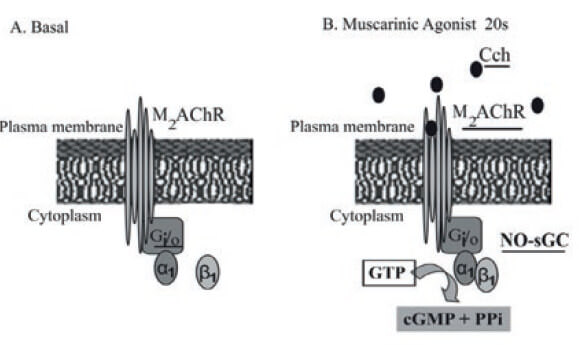
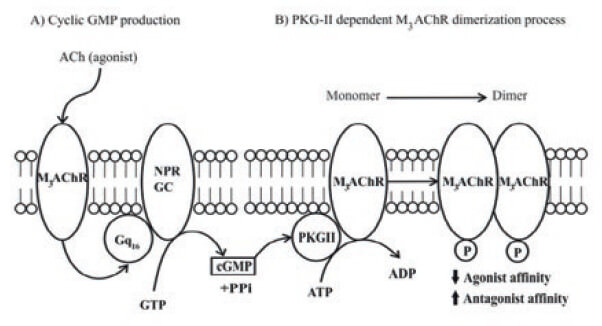
The signal transducing cascade involving the macromolecular complex formed by M3mAChR/ NPR-GC-B, is responsible for the 60s cGMP signal.
This signal cascade was identified at ASM, using biochemical (Lippo de Bécemberg et al., 1995; Alfonzo et al., 1998a; Alfonzo et al., 1998b; Borges et al., 2001; Alfonzo et al., 2006; Bruges et al., 2007) and molecular biology approaches (Borges et al., 2001). The NPR-GC-B is a membrane-spanning homodimer form, which contains an extracellular ligand-binding, trans-membrane, kinase homology, dimerization and carboxyl-terminal catalytic domains (Koller et al., 1991; Alfonzo et al., 2006). NPR-GC-B is activated by CNP, which exists in 22 and 53 amino acid forms that are structurally similar to ANP and BNP (Suga et al., 1992). NPR-GC-B is abundantly expressed in brain, lung, bone, heart and ovary tissue (Nagase et al., 1997). NPR-GC-B contains three intramolecular disulfide bonds and is highly glycosylated on asparagine residues. Moreover, NPR-GC-B is highly phosphorylated and dephosphorylation is associated with receptor inhibition (Potter & Hunter, 1998).
Furthermore, this form, the NPR-GC-B described in BASM, as a novel G-protein coupled NPR-GCB (Lippo de Bécemberg et al., 1995; Alfonzo et al., 1998a), which is present in plasma membranes fractions from this smooth muscle subtype (Lippo de Bécemberg et al., 1995; Alfonzo et al., 1998a). This G protein-coupled NPR-GC-B is located in plasma membranes from BASM shows complex kinetics and regulation. NPR-GC-B is activated by natriuretic peptides (CNP-53>ANP-28) at the ligand extracellular domain, stimulated by Gq-protein activators, such as mastoparan, and inhibited by Gi-chloride sensitive, interacting at the juxtamembrane domain. The kinase homology domain was evaluated by the ATP inhibition of Mn2+-activated NPR-GC-B, which was partially reversed by mastoparan. The catalytic domain was studied by kinetics of Mn2+/Mg2+ and GTP, and the catalytic effect with GTP analogues. These results indicate that NPR-GC-B is a highly regulated nano-machinery with domains acting at cross-talk points with other signal transducing cascades initiated by G protein-coupled receptors and affected by intracellular ligands such as chloride, Mn2+, Mg2+, ATP and GTP. Moreover, this NPR-GC-B from BASM is regulated, in an opposite way by two MAChRs signaling cascades, acting the M2mAChR as an inhibitor and the M3mAChRs as an activator of this NPR-GC-B (Bruges et al., 2007). This novel signal transducing system forms by M3mAChR, a Gq16 being sensitive by mastoparans to activate NPR-GC-B (Alfonzo et al., 2006; Bruges et al., 2007) is illustrated as a model in Figure 5.
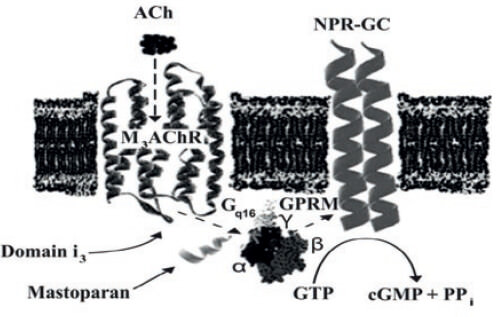
Mastoparans are tetradecapeptides isolated from wasp venom (Hirai et al., 1979; Higashijima et al., 1988), and they are well known as G-protein activators (Higashijima et al., 1990; Shpakov & Pertseva, 2006). These peptides inhibited, in a selective manner, the BASM contraction induced by muscarinic agonist as carbamycholine (Cch) (Hassan-Soto et al., 2012). Interestingly, the biological responses of classic ASM spasmogens as 5-HT (Shi et al., 2007a), was not affected and a potent contractions were displayed (Hassan-Soto et al., 2012).
These findings can be explained since this bioactive amine has been claimed to exert their physiological effects on ASM through specific GPCRs. These receptors are the 5-HT2A, which induced activation of the Gq/11 protein and its downstream effector phospholipase C (PLC) leading to intracellular phosphatidylinositol turnover and Ca2+mobilization (Shi et al., 2007b). These Gq/11 proteins are mastoparan-insensitive ones. The last facts can explain the mastoparan-insensitivity of the serotoninergic transducing cascades at ASM. Furthermore, our results demonstrated that mastoparan inhibits selectively the muscarinic activation without altering other spasmogens transducing cascades at ASM. These experimental results indicate that the most like candidates implicated in mastoparan effects are the heterotrimeric G-proteins coupled to these MAChRs. This mastoparan inhibition on BASM muscarinic activation is mediated through G-proteins altering the generation of the two GMPc signals at 20 s and 60 s under muscarinic agonist exposure (Guerra de González et al., 1999). Mastoparan (50 nM) induced a potent inhibition of these cGMP signals, as shown in Figure 6, that is correlated with a significant reduction on the contractile maximal responses as described (Hassan-Soto et al., 2012). After mastoparan preincubation, the kinetics of cGMP intracellular levels at BASM following the muscarinic agonist exposure were evaluated. Interestingly, the first cGMP signal (20s) decreased in more than 60% and the second signal peak (60s) completely vanished. The significant reduction of 20s cGMP signal peak, which appears at the onset of the ASM contraction, may be explained by the effects of mastoparan on the the M2mAChR/ Go/i protein /NO-sGC signal cascade as above discussed. The dramatic disappearance of the second peak of cGMP (60s) is correlated to the ablation of the muscarinic-dependent ASM contraction. This 60s signal is product of NPR-GC-B (Higashijima et al., 1988; Higashijima et al., 1990; Alfonzo et al., 1998) linked to Gq16 protein activation in isolated BASM plasma membranes fraction (Higashijima et al., 1988) indicates that the signal cascade involving M3mAChR/Gq16/NPR-GC-B is essential for the ASM contractions.
MAChRs are able to induce cGMP signals at BASM as above discussed. The possible existence of a cGMP regulatory feedback mechanism on the MAChR activity in the plasma membranes fractions from BASM was studied. To explore this possibility, plasma membranes fraction was employed, as a simpler and reliable biological system, to evaluate the direct effect of cGMP on the M3/M2mAChRs at the plasma membranes instead of intact ASM or isolated cells, which are a more complicated systems due the plethora of regulatory mechanisms associated with cGMP, as an intracellular second messenger, in a number of physiological functions (Hamad et al., 2003).
The working hypothesis was that muscarinic agonist binds to MAchR inducing its stimulation, which may provoke, some molecular events to guarantee, the activation/termination of these transduction cascades. Plasma membrane possesses some molecular mechanisms for the generation of cGMP linked to activation of MAChRs as above-mentioned. By contrast, to end this muscarinic activation, a MAchR desensitization, via protein phoshorylation has been also described (Hosey et al., 1995; Krupnick & Benovic, 1998; Luo et al 2008).
GPCR phosphorylation-dephosphorylation processes regulate the functional coupling between membrane receptors and G-proteins. Two classes of muscarinic phosphorylations have been reported being the first ones, the G-protein coupled receptor kinases (GRKs) and the G-protein independent kinases phosphorylation. These phosphorylationdephosphorylation processess of MAChRs change the affinity of this receptor for agonists and antagonists. Thus, muscarinic receptor phosphorylation has being reported by GRKs (Hosey et al., 1995; Debburman et al., 1995; Willets et al., 2001), inducing receptor desensitization, which is initiated by the phosphorylation of serine/threonine residues, which promotes uncoupling the receptor from G protein and terminating signaling (Hosey et al., 1995; Krupnick & Benovic, 1998; Luo et al., 2008). Cyclic GMP can affect the MAChR activities at plasma membrane using the [3H]QNB binding activity (Alfonzo et al., 2013). Thus, cGMP, via PKG-II phosphorylates the M3mAChR at plasma membranes from BASM. This PKG-phosphorylation affects its functionality, which is expressed by an increment in the Bmax for [3H] QNB binding activity and displaying a positive cooperativity close to 2.0 for such binding suggesting a M3mAChR dimer formation as shown in Figure 7. Furthermore, a cGMPdependent [32P]-phosphorylation was specific for the M3mAChR using immuno-precipitation assays with anti-M3mAChR (Alfonzo et al., 2015). This novel cGMP biological effect is mediated through the activation of a cGMP-dependent protein kinase (PKG-II) anchored to the plasma membranes fractions from BASM and its existence was also demonstrated (Alfonzo et al., 2013). This [32P]-membrane labeling was affected by muscarinic agonists such as Cch displaying agonistdependent phospho/dephosphorylation reactions. Conversely, 4-DAMP, a selective M3mAChR antagonist, inhibited both the basal and cGMP-dependent membrane protein [32P]-phosphorylations (Alfonzo et al., 2015) supporting the involvement of an unknown PPase. This last possibility is also strengthen by the fact that okadaic acid (OKA), (a protein phosphatase -PPase inhibitor) induced a faster [3H]QNB binding, strengthening the involvement of a membrane-bound PPase (Alfonzo et al., 2013). It has been claimed that M3mAChR dephosphorylation regulates the receptor interactions with G proteins (Wu et al., 2000). The muscarinic receptor signaling regulator, named SET is a PPase 2A inhibitor, which binds to the C-terminal of the i3-loop- M3mAChR (Simon et al., 2006; Simon et al., 2012) decreasing receptor engagement with G proteins. A recent study indicates that the binding site of both SET and PP2A on M3mAChR occurs at the i3-loop (474ITKRKRMS LIKEKKAAQ490). SET specifically binds to the site 476KRKR479 in close vicinity to a domain 484KEKKAAQ490 involved in G protein coupling and activation (Wu et al., 2000; Schmidt et al., 2003) and the domain 480MSLIKEKK485, is the putative phosphorylation site on PKG-II. Thus, this last domain contains the S481, which is located between these two relevant regulatory binding sites, suggesting an important biological function for this PKG-II action.
It is well established that the M3mAChR activation, is the main molecular event, to initiate the ASM contraction. In this sense, to finish the muscarinic activation of ASM, the M3mAChR must be the molecular entity to be knock-off. Thus, the involvement of PKG-II as a cGMP-dependent M3mAChR phosphorylation, is a novel mechanism, presents in ASM cells, to guarantee a feedback control of cGMP on M3mAChR activation. This post-translational reversible modification at M3mAChRs may act as a feedback mechanism to terminate the cGMP dependent muscarinic signal transduction cascades at the sarcolemma of BASM as shown and explained in Figure 7.
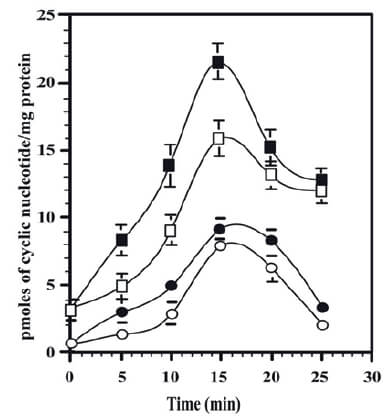
MAChRs are involved in the rise/decline of cAMP/ cGMP levels in ASM (Kamm & Stull, 1989). In isolated BASM strips, a non-selective muscarinic antagonist such as atropine, significantly increased the cGMP and cAMP intracellular levels, in a function of time, being the maximal effects at 15 min, decreasing at longer times, to basal levels in the case of cGMP and remaining higher for cAMP (Mastromatteo-Alberga et al., 2016). This effect was similar to the ones produced by vinpocetine, a specific PDE1 inhibitor (Hagiwara et al., 1984) as shown in Figure 8. The atropine and vinpocetine effects on the cyclic nucleotides (cAMP and cGMP) intracellular levels might be mediated by the inhibition of a cyclic nucleotide phosphodiesterase (Cyclic PDE-1) in BASM as postulated (Mastromatteo- Alberga et al., 2016). Initially, the existence of an Ca2+/CaM dependent PDE1 (CaM-PDE) in intact BASM was demonstrated using an induced cyclic nucleotides accumulation produced by atropine followed by KCl depolarization of BASM strips (Perez & Sanderson, 2005), which produced a fast decline in both cyclic nucleotides levels as the result of CaM-PDE1 activation (Mastromatteo-Alberga et al., 2016). The PDEs are enzymes that hydrolyze cyclic nucleotides (cGMP, cAMP) to the inactive linear form (5-GMP, 5-AMP), thereby regulating both the duration and the amplitude of cyclic nucleotide signaling. It is possible that KCl induces membrane depolarization and initiates a Ca2+ influx, through the opening of L-type Ca2+channels located at BASM sarcolemma increasing the [Ca2+]i and the active CaM levels (Perez & Sanderson, 2005) producing the activation of the BASM contraction machinery (Simon et al., 2006) and the PDE1s (Sharma et al., 1980; Charbonneau, 1990).
It has been reported that ASM exhibits Ca2+/CaM dependent PDE1s (CaM-PDE 1) that hydrolyze cAMP and cGMP with equal capacity (Beavo, 1988; Torphy & Cieslinski, 1990; Shahid et al., 1991). This is the main reason to consider this PDE 1 family as the target of atropine and vinpocetine actions because both drugs increased the cAMP and cGMP levels as discussed.
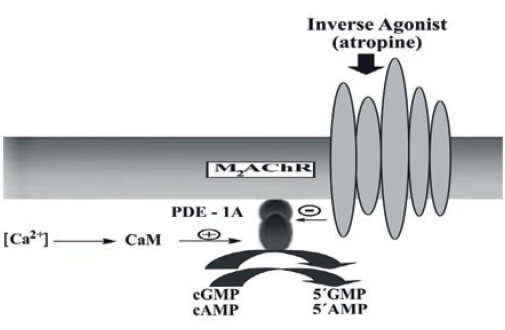
CaM-PDEs are mediators between the Ca2+and cyclic nucleotides second messenger allowing cyclic nucleotide-dependent processes to be regulated by [Ca2+]i (Beavo, 1995) PDE1 family has three genes (PDE1A, PDE1B, and PDE1C) (Beavo, 1995; Sonnenburg et al., 1995). This PDE1 from BASM was identified as a vinpocetine and atropine-sensitive PDE being located at plasma membranes of this smooth muscle subtype (Mastromatteo-Alberga et al., 2016). At this plasma membrane fraction, the muscarinic receptor involves, in this muscarinic antagonist inhibition, was identified as an M2mAChR (Misle el al., 1994; Misle et al., 2001) as shown in Figure 9. This PDE1 was inhibited by muscarinic antagonists acting on M2mAChRs at the sarcolemma from BASM (Mastromatteo-Alberga et al., 2016). Thus, methoctramine (M2mAChR selective antagonist) (Michel & Whiting, 1988) was more effective than 4-DAMP (an M3mAChR selective antagonist) (Michel et al., 1989) inhibiting this membrane-bound PDE1A. The inhibitory concentration IC50 for methoctramine, ( IC50 = - 8.12 ± 0.21) and for 4-DAMP, (IC50= - 6.68 ± 0.13). This pharmacological profile belongs to an M2mAChR (Misle et al., 1994). ASM displays a mixed population of M2 and M3 subtypes (Eglen et al., 1996) being the M2 subtype, the most abundant at BASM (Misle et al., 1994; Misle et al., 2001). In addition, the isolation and purification of this plasma membrane-bound PDE1 was characterized as a vinpocetine-inhibited and CaM-stimulated PDE activity (Sharma et al., 1980) with MW of 58 kDa and identified by Western blotting as PDE1A as described (Mastromatteo-Alberga et al., 2016).
It is well established that M2mAChRs participate directly in ASM contraction, via Gi/o proteins (Jones et al., 1987; Sankary et al., 1988; Fernandes et al., 1992; Nakahara et al., 2002; Zhou et al., 2008; Semenov et al., 2011; Pláceres-Uray et al., 2010a). However, our experimental data provided by the muscarinicantagonists inhibition of PDE1A at plasma membranes from BASM, which took place in the absence of GTP or analogs, suggesting a direct coupling mechanism between M2mAChR and this PDE1A (Mastromatteo- Alberga et al., 2016). In this sense, a direct coupling mechanism between the M2AChR and the target enzyme has been demonstrated for other GPCR, as a direct interaction between the M3mAChR, and its effector enzyme PLC β3 (Kan et al., 2014). All these experimental evidences support a model for the interaction of M2mAChR and PDE1A illustrated in Figure 10.
In summary, we provide evidence that the molecular signal transducing functional machinery named “signalosomes”, one comprising by M3mAChRs, NPRGC- B, PKG-II and the second ones, the M2mAChRs linked to cGMP-PDE 1A, which are located at the plasma membrane of BASM (Mastromatteo-Alberga et al., 2016).
![Figure 9.
Effect of muscarinic antagonists on the PDE1 activity at plasma
membranes fraction. Plasma membranes from BASM (20–30 μg
membranes protein) fractions were incubated with [3H] cGMP as PDE
substrate as described (Mastromatteo-Alberga et al., 2016). A dose
titration curve of muscarinic antagonists such as methoctramine (□)
and 4-DAMP (▲) were performed. Data are the mean ± SE of three (3)
different plasma membranes from BASM preparations. Taken from
(Mastromatteo-Alberga et al., 2016).](9.jpg)
The airway structural changes play an important role in the development of the airway hyperresponsiveness presents in asthma. Furthermore, the remodeling of the airways is a relevant process in asthma, being the most important feature the hyperplasia (proliferation) of smooth muscle (Gosens et al., 2007; Kistemaker et al., 2012). The role of these MAChRs and their signal transducing cascades in airway smooth cell proliferation studies were performed in rat ASMC. Initially, we explored the effect of muscarinic agonist, carbamylcholine (Cch) on cell proliferation as described (Pláceres-Uray et al., 2010a). Fetal Bovine Serum (FBS) increased rat ASMC proliferation, being significant at 48 and 72 hr as shown in Figure 11. However, muscarinic agonist Cch, in a dose dependent manner, decreases the ASMC proliferation induced by 10% FBS (Figure 11). Thus, a dose dependent inhibitory effect on ASMC proliferation by Cch was significant at 48 and 72 hrs. Proliferation inhibition was not due to death cell because ASMC viability (90-95%) in presence of Cch was confirmed with blue dye exclusion method (Pláceres-Uray et al., 2010b).
Trying to understand this novel finding on the ability of muscarinic agonist (Cch) to exhibit antiproliferative properties especially at high mitogen concentration (10% FBS); the identification of the mAChR involves in this inhibitory effect was relevant. Thus, a pharmacological approach was performed using selective muscarinic antagonists, such as 4-DAMP (for M3mAChR) and AF-DX-116-DS (for M2mAChR). It was found, this anti-mitogenic Cch effect was reversed, in a dose dependent manner, by the selective muscarinic antagonists as AF-DX- 116 (M2mAChR antagonist) (Gosens et al., 2004), which was more efficient than 4-DAMP (M3mAChR antagonist) (Michel et al., 1989) to reverse this novel Cch inhibition activity as shown in Figure 12. The proliferative stimulatory responses displayed by muscarinic antagonists reversed significantly the anti-mitogenic Cch (1x10-5M) action (p < 0.05). Thus, the log IC50 ± SE for AFDX-116-DS = −9.40 ± 0.37 and 4-DAMP = −7.11 ± 0.71 were estimated. The difference between these log IC50 values is more than 2 orders of magnitude, that support a pharmacological profile of AFDX 116 > > 4-DAMP, which clearly belongs to an M2mAChR (Misle et al., 1994; Misle et al., 2001).
![Figure 11.
Rat ASMC proliferation responses to muscarinic agonist [Cch] concentrations.
ASMC (1 × 103 cells/well) were cultured in 96 wells plates
with increasing concentration of muscarinic agonist Cch in medium
without FBS during 48 (□), 72 (Δ), and 10% FBS, during 48 (■), y 72 (▲)
hrs. Cell proliferation was determined using a colorimetric method
(MTS-PMS), measuring O.D at λ = 492 nm. Data are the mean ± SE of
n = 6 experiments for triplicate. Mitogen responses at 48 and 72 h by
10% FBS vs. basal was significant (*) p < 0.05. The inhibitory effect of
Cch (-7,-5,-3) vs 0 Cch was significant (**) p < 0.05.](11.jpg)
In normal rat ASMC, the exposure to 10% FBS, produces a mitogen-induced proliferation, which was inhibited by muscarinic agonist, via an M2mAChR as above discussed. It is well known that asthma and COPD exhibit an “inflammatory environment”, rich in ASMC mytogens (Barnes, 2008). Interestingly, it has been reported that in murine (BALB/c mice) models of chronic asthma, sensitized and challenged to ovalbumin, the expression of the M3mAChR was inhibited and M2mAChR was elevated by the administration of tiotropium bromide (Kang et al., 2012). It is well known that anticholinergic drug (tiotropium bromide) (Kang et al., 2012; Busse et al., 2016),which selectively antagonizes the M3mAChR subtype, could be beneficial in attenuating airway remodeling in chronic asthma. These findings on the ability of M2mAChR to be an anti-mitogenic receptor, can explain these experimental findings of tiotropium bromide (Kang et al., 2012) and open new avenues for drug design to treat these chronic inflammatory respiratory diseases such as asthma and COPD (Kang et al., 2012; Busse et al., 2016; Kang et al 2012).
The molecular mechanisms involved in these novel M2mAChR anti-proliferative effects are unknown. However, the involvement of M2mAChR, in molecular mechanisms, related to cell growth or regression has been claimed (Nicke et al., 1999).
It has been demonstrated that the activation of M2mAChR inhibits cell growth and survival in human glioblastoma cancer stem cells. (Alessandrini et al., 2015). In urothelial bladder cancer cells similar findings have been reported (Pacini et al., 2014). This muscarinic agonist inhibition mechanism of ASMC proliferation may be the result of the cGMP/PKG activation cascade signaling pathways (Gilotti et al., 2014). It is our interest the cGMP/PKG transducing cascade, in BASM. It is known that muscarinic agonists (Cch) rise the cGMP levels generating two cGMP signals, the first (20s) as a product of M2mAChR /NO-sGC coupling mechanism described (Guerra de González et al., 1999). This NO-sGC is an enzymehighly expressed in the lungs (Papapetropoulos et al., 2006). However, it has been claimed that excessive NO production that occurs in asthma induces a down-regulation of NO-sGC (Pláceres-Uray et al., 2010b; Papapetropoulos et al., 2006). The second signal cGMP (60s), is produced, via the M3mAChR /Gq16 protein/NPR-GCB cascade (Lippo de Bécemberg et al., 1995; Alfonzo et al 1998a; Alfonzo et al 1998b; Borges et al., 2001; Alfonzo et al., 2006; Bruges et al., 2007). Thus, the M2mAChR/Go/i /NO-sGC and the M3mAChR /Gq16 protein/NPR-GC-B cascades produce cGMP, which can activate the cyclic GMP-dependent protein phosphokinases (PKG), which can exist as two isoenzymes termed PKG-I and PKG-II (Lohmann et al., 1997; Francis et al., 2010). These PKGs can phosphorylate transcription factors associated with inhibition of gene expression that promote cell cycle, also induce increment of proteins that leads cell cycle arrest as p21Cip1/Waf1 converting the PKG, as an anticancer promising signal cascades (Cen et al., 2008). In this sense, it has been claimed that PDE5 inhibitors, that increased the cGMP levels, have anticancer activities express as anti-proliferative mechanisms in multiple carcinomas. Thus, these compounds reduced the protein expression of cyclin D1, while p21 protein expression was increased. Furthermore, it has been found that PKG Iβ being responsible for the antiproliferative effects (Ren et al., 2014).
It is possible that these signal cascades may be involved in this anti-mitogenic effect of M2mAChR. Actually, the specific molecular mechanisms linked to the anti-mitogenic M2mAChR responses are under intense research.
It has been claimed that a dysfunction of the MAChR signal transducing cascades has been implied in the pathophysiological mechanisms of asthma (Gosens et al., 2004; Racke & Matthiesen, 2004; Belmonte, 2005; Gosens et al., 2006; Gosens et al., 2007; Kistemaker et al., 2012) and Chronic Obstructive Pulmonary Disease (COPD) (Racke & Matthiesen, 2004; Gosens et al., 2006). The pathophysiological relevance of the signal transducing cascades here described as the M2mAChR/NOsGC and the M3mAChR/NPR-GC-B, were evaluated in ASMC from a murine (rat) chronic asthma model (Vanacker et al., 2001). This is a murine experimental model of allergic asthma, is induced by long exposure to ovalbumin (OVA), which mimics several of the central hallmarks of human bronchial asthma including airway hyperresponsiveness and immune cell lung infiltration (Vanacker et al., 2001).
The muscarinic receptor (M3mAChR/M2mAChRs) expression levels in ASM determined by radioligand binding were not significantly different from normal and asthmatic patients or animal asthma models. These facts conduce to evaluate the guanylylcylases activities linked to these receptors, by the cGMP production, which may relevant to ASMC proliferation. Accordingly, the NOsGC linked to M2mAChR and the NPR-GC-B associated to M3mAChR were determined, in CD rat ASMC from CONTROL and a murine experimental model of allergic asthma induced by long exposure to ovalbumin (OVA).
Cultured Airway Smooth Muscle Cells (ASMC) from CONTROL and OVA-CD rats were used to determine the cGMP production under the exposure to muscarinic agonists (Cch) (Alfonzo et al., 1998a; Alfonzo et al., 1998b; Guerra de González et al., 1999; Bruges et al., 2007), an inhibitor of NO-sGC (ODQ) (Wedel et al., 1995; Pláceres-Uray et al., 2010b; Pláceres-Uray et al., 2011; Derbyshire & Marletta, 2012; Russwurm et al., 2013) and the natriuretic peptide (CNP) to stimulate the NPR-GC-B (Koller et al., 1991; Borges et al., 2001; Alfonzo et al., 2006; Bruges et al 2007).
It can be seen in Figure 13 that OVA-ASMC showed low basal cGMP production compared to CONTROL ASMC as previously reported (Pláceres-Uray et al., 2010b). Thus, these basal GC activities were, more than 50% sensitive to ODQ, indicating that NO-sGCs are present in these ASMC. It has been reported that the bronchoconstriction observed in asthma is accompanied by changes in NO-sGC activity possibly due to substantial reduced sGC expression reflected in decrease the steady state levels of sGC subunit mRNAs and protein level expression, as found in intact lung tissue from OVA-sensitized mice (Papapetropoulos et al., 2006).
The muscarinic agonist (Cch) increased the cGMP intracellular levels in all ASMC, which were also inhibited more than 50% by ODQ. Specifically, in the OVA-ASMC, CCh increased 3.5 fold the GC activity being 1.5 fold in the CONTROL-ASMC. These results in ASMC confirmed all previous experimental findings on the ability of muscarinic agonists to increase the cGMP intracellular levels as reported in isolated ASM strips (Katsuki & Murad, 1977; Guerra de González et al., 1999). Interestingly, CNP (activator of NPR-GC-B) stimulate (> 2 fold) the GC activity in CONTROL ASMC while in the OVA ASMC was > 3.0 fold. However, ODQ potentiated the CNP activation in the OVA-ASMC suggesting that NO-sGC may regulate the NPR-GC-B.
A relevant finding in this work was that CNP and Cch act synergistically, and this combination displays the maximal GC activity in cultured ASMC. Thus, this synergistic effect was more significant (> 6.0 fold) on the OVA-ASMC. In addition, ODQ inhibited this CNP plus CCh effect, in about 25% (NO-sGC), in both cells types (Figure 13). One possible explanation for this synergistic effect is related to the property of the G-protein coupled NPRGC-B nano-machine described in BASM, that possess an intracellular domain named as the G-protein regulatory module (GPRM) (Alfonzo et al. 2006), in which, molecular components or events coming from the M3mAChR/Gq16 activated cascade by muscarinic agonist (Cch), can potentiate or facilitate the CNP activation of the NPR-GC-B, which occurs at the extracellular domain (Koller et al., 1991; Suga et al., 1992; Borges et al., 2001; Alfonzo et al., 2006; Brown et al., 2013).
Due to the important finding that the Cch and CNP act synergistically, the M3mAChR/G-protein/NPR GC-B cascade on the ASM was profoundly studied (Lippo de Bécemberg et al., 1995; Alfonzo et al., 1998a; Alfonzo et al., 1998b; Borges et al., 2001; Alfonzo et al., 2006; Bruges et al., 2007). Consequently, the effect of several activators of this G-protein coupled NPR-GC-B such as natriuretic peptides (CNP) and mastoparans (G-protein activator specially Gq16) were evaluated as previously described in BASM (Lippo de Bécemberg et al., 1995; Alfonzo et al., 1998a; Alfonzo et al., 1998b; Borges et al., 2001; Alfonzo et al., 2006; Bruges et al., 2007; Gosens et al., 2007).
It can be seen in Figure 14 that the CNP stimulated GC activity was more than 3 times in OVA-ASMC and 1.5 times in Control-ASMC indicating that NPR-GCB was present in Control and OVA-ASMC, which are similar behavior described in plasma membranes preparations from ASM cells (Lippo de Bécemberg et al., 1995; Alfonzo et al., 1998b; Alfonzo et al., 2006). The muscarinic agonist (Cch) increased in 1.3 times, the Control ASMC GC activity while increased 2.2 times in OVA-ASMC. Thus, the OVA-ASMC in comparison to Control ASMC showed a more significant stimulation by these NPR-GC-B activators. Interestingly, the combination of (Cch plus CNP) increased in more than 6 times the OVA-ASMC-GC activity being only 1.5 times in Control ASMC. As expected, mastoparan, an activator of NPR-GC-B, via Gq16 (Bruges et al., 2007; Hassan-Soto et al., 2012) dramatically increased in 6 times, the OVA-ASMCGC activity and 1.5 times in Control-ASMC (Alfonzo et al., 2006; Bruges et al., 2007). However, at OVAASMC, Cch did not affected the hyperstimulation induced by mastoparan. However, the combination of CNP and mastoparan rendered in a reduction of the NPR-GC activation at OVA-ASMC These data showed for the first time that there is a hyperstimulation of M3mAChR/Gq16/NPR-GC-B cascade in ASMC from an experimental asthma model (Pláceres-Uray et al., 2011), which support the classic statement that AC mediates airway smooth muscle (ASM) contraction primarily the activation of M3mAChRs (Roffel et al., 1988; Eglen et al., 1996; Misle et al., 2001; Belmonte, 2005; Gosens et al., 2006; Brown et al., 2013).
Taking together all these results indicate that the ASMC from OVA-sensitized rats express a reduced NO-sGC activity and a hyperstimulation of M3mAChR/Gq16/NPR-GC-B cascade. The imbalance between these two signal transducing cascade, one of them regulated by M3mAChR/Gq16/NPR-GC-B cascade can contribute to airway hyperreactivity to muscarinic agonists and it might be implicated in the Ach linked hyperplastic and remodeling smooth muscle responses present in asthma and COPD (Kistemaker & Gosens, 2015).
Consequently, this work opens new pharmacological and therapeutical approaches for the treatment of these chronic respiratory diseases, such as asthma and COPD, in which, the ASM is involved.
This review discussed the recent experimental findings about the role of muscarinic receptors and in regulating the function of cGMP at ASM, in normal and “pathological” conditions as asthma. The population ratio of M2mAChRs/ M3mAChRs at ASM is 4:1. This fact may help to understand the existence of three “muscarinic signal transducing signalosomes”: One comprised by M3mAChR/Gq16/NPR-GC-B; PKG-II located in the plasma membrane of ASM and the second ones formed by M2mAChRs, NO-sGC and transient location at the plasma membrane of ASM and the third, the novel M2mAChRs coupled to PDE1A. These three signalosomes regulate a “sarcolemmaassociated cGMP pool” (Caulfield, 1993; Challis et al., 1993 ; Kostenis et al., 1999; Wessler & Kirkpatrick, 2001; Proskocil et al., 2004; Oldham & Hamm, 2008; Wessler & Kirkpatrick, 2008) as second messenger, which streams down to activate a membrane-bound PKG-II, which then phosphorylates the M3mAChR at the i3-loop extending from Thr450-Q490, inducing the desensitization of this M3mAChR subtype, in a feedback mechanism at plasma membrane level (Lucchesi et al., 1990). The M3mAChR, a prototypic class A GPCR, which preferentially couples to the family of G proteins, is involved in numerous important physiological functions in ASM. Interestingly, M3mAChR in ASM, is involved in the cholinergic tone, which contributes to airflow obstruction and chronic airway inflammation in asthma and COPD. It is known that anticholinergics are effective bronchodilators by blocking this M3mAChR subtype but also increased the expression of M2mAChR (Shi et al., 2007a).
In the other hand, the M2mAChRs, NO-sGC “muscarinic signal transducing signalosome is involved in the generation of cGMP signal at the plasma membrane level, which is independent of the presence of NO, classic activator of this NO-sGC. Moreover, M2mAChRs is linked to two novel signal transducing signalosomes, one involves the direct coupling of this receptor subtype to a PDE1A, to hydrolyze the cGMP, and in this way to end this second messenger. The last one, is an unknown antimitogenic signal cascade that connect M2mAChRs to the nuclear machinery probably the regulation of the intracellular signaling pathways: 1) The cGMP/ PKG activation cascade, and 2) MAPK activation: p38 MAPK and JNK cascade to control the nuclear machinery by blocking the cell cycle and in this way, act as anti-mitogenic in the ASM cells, which is relevant process in the airway remodeling presents in asthma and COPD. The molecular components of the last signal cascade are unknown.
Thus, understanding, how the M3mAChR and M2mAChRs work at the molecular level is of considerable relevance for designing novel classes of drugs that can modulate M3mAChR and M2mAChR functions, which may be significant for therapeutic purposes in pathological conditions such as asthma and COPD.
ASM: Airway Smooth Muscle, ODQ:(1H-[1, 2,4] Oxadiazolo[4,3-a]quinoxalin-1-one), COPD: Chronic Obstructive Pulmonary Disease, M3mAChR: muscarinic receptor subtype 3, and M2mAChR: muscarinic receptor subtype 2, PDE: Cyclic nucleotide phosphodiesterase, cGMP: cyclic Guanosine monophosphate, PKG: cGMP dependent protein kinase, NPR-GC: Natriuretic Peptide sensitive guanylyl cyclase type B, NO-sGC NO sensitive soluble guanylyl cyclase, Cch Carbamylcholine, GPCR G protein coupled receptor, Gq16 Heterotrimeric G q16 protein.
The authors declare no conflict of interest.
This work was supported by a grant from CDCHUCV # PG 09-8629-2013/2 (to ILB). The authors thank Dr Coral Wynter for revision of the English version of this manuscript.