
La incidencia de demencias se espera aumente de forma alarmante en los próximos años en los países de Latinoamérica y Venezuela no escapa a esta realidad. Existen formas de demencia con herencia autosómica dominante como la Demencia Fronto Temporal (DFT) y la Enfermedad de Alzheimer (EA) de aparición temprana, en donde tienen gran validez los estudios genéticos para determinar que sujetos van a presentar la patología. La EA de aparición tardía, es la forma de demencia más común y existen variantes genéticas que pueden identificar individuos de alto riesgo. Un ejemplo lo constituye el alelo e4 de gen que codifica para APOE. Sin embargo, la sola presencia de esta u otras variantes genéticas no son determinantes para la aparición de la EA. Por esta razón las mismas deben ser analizadas con cautela en la población general. El estudio de estas variantes se hará cada vez más relevante en la medida que se desarrollen fármacos capaces de prevenir y/o detener la historia natural de la enfermedad
Palabras clave: Alzheimer; variantes genéticas; riesgo; prevención; APOE e4.
The incidence of dementia is expected to increase dramatically in the coming years in Latin America, and Venezuela does not escape this reality. There are forms of dementia with autosomal dominant inheritance such as Fronto Temporal Dementia (FTD ) and early onset Alzheimer‘s Disease (AD). Genetic studies for these conditions in order to determine affected subjects are of relevance. Late-onset AD is the most common form of dementia and there are genetic variants that can identify high-risk individuals. The best example is the e4 allele of the APOE gene. However, the mere presence of this or other genetic variants are not conclusive for the development of AD. Therefore their analysis should be undertaken with caution in the general population. Variant determination will become increasingly relevant as soon as drugs able to prevent and / or stop the natural history of disease are developed.
Key words: Alzheimer; genetic variants; risk; prevention; APOE e4.
La Organización Mundial de la Salud (OMS) define la demencia como un síndrome crónico caracterizado por el detrimento progresivo de la función cognitiva. La demencia repercute en la memoria, el pensamiento, la orientación, la comprensión, el cálculo, la capacidad de aprendizaje, el lenguaje y el juicio. El deterioro generalmente ocurre en edades avanzadas, observándose en personas mayores de 60 años y es lo suficientemente discapacitante para reducir de manera significativa la habilidad de los individuos para realizar las actividades cotidianas. El ICD-10 (“International Statistical Classification of Diseases and Related Health Problems”) de la OMS clasifica las demencias en cuatro grandes grupos: a) Demencia en la Enfermedad de Alzheimer (EA), b) Demencia Vascular, c) Demencia en otras enfermedades que incluye la Demencia Fronto-Temporal (DFT) y d) Demencias no especificadas.
Una de las entidades dentro del grupo de las demencias que ha sido considerablemente estudiada desde el punto de vista genético es la DFT, esta se caracteriza por la degeneración progresiva de los lóbulos temporal y frontal o ambos. Desde el punto de vista clínico hay un deterioro progresivo del comportamiento, el lenguaje, afectándose en un menor grado la memoria y la función visuoespacial (Neary et al., 1998). La DFT constituye la segunda causa más común de demencia de inicio temprano (antes de los 60 años), después de la EA. Mutaciones en tres genes explican más del 80% de los casos de DFT con patrón hereditario autosómico dominante. Estos genes son el MAPT, que codifica para la proteína Tau, estabilizadora del citoesqueleto neuronal; el gen GRN, que da origen a la proteína granulina que regula el crecimiento celular y el gen C9ORF72, el cual genera una proteína del mismo nombre, muy abundante en las células nerviosas.
Por su parte, la EA es la forma más frecuente de demencia, representando un 60 a un 70% de los casos. Las características principales de esta patología, desde el punto de vista anatomo-patológico, lo constituyen una pérdida neuronal severa, agregación de proteína beta amiloide en placas seniles extracelulares y formación de ovillos neurofibrilares constituidos por proteína Tau hiperfosforilada. La predisposición genética a la EA ha sido ampliamente estudiada, por lo que va a constituir el enfoque principal de este trabajo y la discutiremos más adelante.
Las cifras del reporte de Alzheimer mundial de 2015 indican que para el año pasado 46 millones de personas habían sido diagnosticadas con demencia. Solo en el 2015 se reportaron 9,9 millones de casos nuevos en el mundo. El reporte estima un aumento importante en este número que pudiera llegar a 131,5 millones de pacientes en el año 2050. Los datos también sugieren que 58% del total de personas que padece esta enfermedad vive en países clasificados por el Banco Muncial como de bajo o medios ingresos. En estos países la proporción de personas con demencia va a aumentar a 63% para el año 2030 y a 68% para el año 2050. Estos números causan preocupación, sobre todo si se analizan las proyecciones específicas para nuestro continente y nuestro país. Se calcula para Latinoamérica un incremento porcentual de 393% de personas que padecerán EA en un periodo aproximado de 40 años (2001-2040), el más alto del mundo (Reitz et al., 2011). En Venezuela 130.000 adultos mayores (7 de cada 10) sufren de la patología (Fundación Alzheimer Venezolana). La mayor incidencia se reporta en el estado Zulia 12,13%, seguida del estado Miranda 11,67% y luego el Distrito Capital 9,91%. El número de casos aumenta exponencialmente con la edad, de hecho dentro de los países estudiados por el grupo 10/66 “Dementia Research Group” (Cuba, República Dominicana, Perú, México, China y Venezuela), Venezuela tuvo la mayor incidencia dentro del grupo de personas mayores de 80 años (Prince et al., 2012). Esto es de gran relevancia, dado que las proyecciones del Instituto Nacional de Estadística (INE), sugieren que la población de adultos mayores se va a triplicar en 30 años, pasando de 1.991.738 en el año 2015 a 6.304.070 en el año 2045. Este aumento en la población de adultos mayores va entonces a incidir directamente en las cifras de demencia del país, a menos que se puedan desarrollar medidas preventivas eficaces, como el diagnóstico y tratamiento precoz, a fin de detener el avance de esta enfermedad.
La EA se clasifica en Alzheimer precoz y Alzheimer de aparición tardía (clásica). En el primer caso los síntomas aparecen antes de los 65 años, mientras que en la EA de aparición tardía se diagnostica en mayores de 65 años. La edad y el sexo femenino constituyen los factores de riesgo mejor caracterizados para padecer la EA clásica. Según el Instituto Nacional del envejecimiento (National Institute of Aging), el riesgo de desarrollar EA se duplica cada 5 años luego de alcanzar los 65 años.
Es de hacer notar, que en los últimos años la búsqueda de factores genéticos como predictores de riesgo para padecer EA se ha constituido en un gran campo de investigación. El hallazgo de mutaciones con penetrancia completa en genes que codifican proteínas como la proteína precursora de amiloide (APP), presenilina 1 (PSEN1) y presenilina 2 (PSEN2) como causantes de EA familiar precoz autosómica dominante, ha sido un hito muy relevante. En la base de datos de mutaciones para la EA y DFF (http://www.molgen.ua.ac.be/admutations) se han reportado 49 mutaciones en APP, 216 en PSEN1 y 15 en PSEN2.
Uno de los grupos con EA familiar mejor identificados en nuestras latitudes, es el de los pobladores de la población de Yumaral del Departamento de Antioquia, Colombia. El Dr. Francisco Lopera ha estudiado un grupo de cerca de 5.000 personas pertenecientes a 25 diferentes familias con la mutación E280A en PSEN1, incluyendo a individuos sanos y en situación de riesgo. En este grupo de individuos la edad media de inicio es de 46,8 años (34-62), la duración media de la demencia es de diez años, con una edad promedio de fallecimiento a los 54,8 años (38-64). No reportaron diferencias significativas entre mujeres y hombres (Alzheimer’disease collaborative group). La existencia de este conglomerado ha sido de gran ayuda para la comunidad científica, e investigadores de diversas latitudes se han abocado a estudiar a los habitantes de Yarumal, a fin de tratar de establecer un tratamiento precoz que detenga la aparición y/o progresión de la patología. En estos casos las pruebas genéticas contribuyen como herramienta diagnóstica, para detectar los individuos a riesgo antes de que aparezcan las características clínicas de la EA.
En nuestro laboratorio, hemos buscado esta misma mutación (E280A en PSEN1) en pacientes con Alzheimer precoz, sin embargo no hemos tenido éxito en los pocos pacientes estudiados.
Por otra parte, el factor de riesgo más importante desde el punto de vista genético para desarrollar EA clásica es la presencia del alelo e4 del gen que codifica para la Apolipoproteína E (APOE). Individuos con un alelo e4 tienen una probabilidad 3 veces mayor de desarrollar Alzheimer que aquellos que no lo poseen y los portadores homocigotos para el e4 tiene 15 veces más posibilidad de padecer la enfermedad (Saunders et al., 1993). El alelo e4 constituye también un factor de riesgo para la ateroesclerosis (Zhu et al., 2016), la angiopatía amiloidea cerebral (Shinohara et al., 2016), la demencia con cuerpos de Lewy (Tsuang et al., 2013) y un factor que incrementa el deterioro cognitivo en pacientes con esclerosis múltiple (Shi et al., 2011). Estas observaciones sugieren que el alelo e4 debe estar asociado con una acelerada neurodegeneración común a varias enfermedades neurodegenerativas. Es conveniente aclarar que la sólo presencia de este alelo no significa que el individuo va a sufrir la enfermedad, únicamente indica que tiene un riesgo mayor de padecerla. De hecho, alrededor del 75% de las personas portadores de una copia del alelo e4 nunca manifiestan la patología. Por otro lado, 50% de las personas de padecen EA no tienen el alelo de alto riesgo (Ferrer et al., 1995). Es por esto que el tamizaje de pacientes en los que se estudie la predisposición genética para EA, debe ser realizado por personal especializado que sepa explicar detalladamente el resultado de los análisis genéticos que se llevan a cabo en el paciente.
Nuestro grupo de investigación ha venido realizando estudios en este sentido en adultos mayores de Caracas, aparentemente sanos y con diagnóstico de EA. Nuestros datos preliminares demuestran que la frecuencia alélica en nuestra población control es comparable con lo reportado para la población caucásica a nivel mundial y en Maracaibo, Venezuela (Tabla 1). Estamos en este momento analizando los grupos de pacientes.
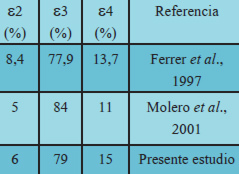
También hemos tenido la oportunidad de analizar una población autóctona venezolana de Pemones Kamarokotos, habitantes del Valle de Kamarata, Edo. Bolívar, donde el hallazgo más relevante ha sido la ausencia del alelo e2. Estos hallazgos coinciden con los reportados en otras poblaciones indígenas Venezolanas (Fernández-Maestre et al. 2005).
Recientemente se han descrito por lo menos 21 loci de riesgo adicionales en estudios de asociación del genoma ampliado (GWAS) y plataformas de secuenciación masiva (Van Cauwenberghe et al., 2016). Estos genes están involucrados en el metabolismo de lípidos, la respuesta inmune, endocitosis y variantes que alteran la función sináptica, entre otros (Karch et al., 2014). Sin embargo cada uno de estos loci sólo contribuye en un pequeño porcentaje a la susceptibilidad para EA. Por lo tanto todavía, una gran proporción de la herencia genética para EA no ha podido ser explicada por GWAS.
En los últimos años, se ha identificado que la variante R47H en el gen TREM2 (receptor de activación expresado en células mieloides 2) tiene una asociación importante con la EA (Jonsson et al., 2013). Dado al papel antiinflamatorio de la proteína TREM en el cerebro, se sospecha que de alguna forma interfiere con la habilidad de este órgano para prevenir la formación de las placas amiloides (Hickman y El Khoury, 2014). Los individuos con esta variante tienen 3,4 veces más riesgo de presentar EA. Nuestro laboratorio también está analizando esta variante en la población venezolana.
El costo emocional y económico de este tipo de patologías es muy grande, especialmente para los familiares que están al cuidado de la persona enferma. De hecho un paciente con alteraciones cognitivas severas necesita cuidados constantes la mayor parte del tiempo. Se ha estimado que “el cuidador” tiene un riesgo de 3 a 5 veces mayor que la población general de sufrir depresión y los gastos de manutención del enfermo (hospitalización, tratamiento farmacológico, etc.) son excesivos. Es por ello, que es fundamental la detección precoz de factores de riesgo y la pesquisa de biomarcadores de dicha patología en nuestra población a fin de practicar estrategias de prevención temprana, con la finalidad de retardar la aparición de la enfermedad y de ser posible, modificar su historia natural. Lamentablemente en la actualidad, las opciones terapéuticas para EA son limitadas y poco efectivas.
Los estudios genéticos en aquellos pacientes con riesgo familiar importante de demencia y cuando se sospecha de un síndrome hereditario, como EA o DFT autosómico dominante, están plenamente justificados. Sin embargo, para EA clásica sigue siendo un debate, ya que solo se pueden estudiar genes de susceptibilidad. Todavía se considera a APOE e4 como el factor de riesgo de más relevancia desde el punto de vista genético para EA clásica.
Se han ensayado drogas específicas para la enfermedad basadas en la predisposición genética, incluyendo la inmunoterapia con bapineuzumaban, un anticuerpo específico contra el segmento N terminal de la proteína Ab que había sido efectivo para prevenir la deposición del Ab en los portadores de APOE que presentaban enfermedad leve o moderada (Liu et al., 2013). Sin embargo, en los estudios de fase III, no se han obtenido los resultados deseados con este medicamento (Vandenberghe et al., 2016). En estos momentos se están llevando a cabo estudios específicos que buscan transferir la secuencia codificante de la APOEe2 (alelo protector) al sistema nervioso central de individuos de alto riesgo para desarrollar Alzheimer (APOEe4 positivos), usando transferencia con Virus Adeno Asociados (Alzheimer´s drug discovery foundation). Aún se está en espera de los resultados de este tipo de ensayos.
A medida que aumente la disponibilidad de fármacos para pacientes con predisposición genética se justificará cada vez más el uso de pruebas genéticas para la identificación y tratamiento precoz de los pacientes a riesgo.